Zum Inhalt springen
Zum Inhalt springen Die Phase des Zulassungsverfahrens für neue Arzneimittel (NDA) ist oft die Zeit, in der Pharma-Teams den größten Druck verspüren. Es ist der Zeitpunkt, an dem jahrelange Entwicklung, Tests und Investitionen formell von der FDA geprüft werden.
Vereinfacht ausgedrückt ist ein NDA (New Drug Application) in der Pharmabranche das Dokument, das ein Unternehmen bei der FDA (Food and Drug Administration) einreicht, um die Zulassung eines neuen Medikaments zu beantragen. Dies ist ein wichtiger Meilenstein in jedem Arzneimittelentwicklungsprozess.
Wenn Sie also eine neue Rezeptur entwickeln oder die Produktion ausweiten möchten, müssen Sie den Ablauf des NDA-Einreichungsverfahrens verstehen. Genau darauf konzentrieren wir uns heute. Legen wir los.
Was ist eine NDA in der Pharmabranche?

Eine Geheimhaltungsvereinbarung ist die offizielle Anfrage an einen Pharmaunternehmen Der Antrag auf Zulassung eines neuen Medikaments bei der FDA ist der letzte Schritt im Arzneimittelentwicklungsprozess und umfasst alle Unterlagen, die die FDA benötigt, um über die Sicherheit und Wirksamkeit des Medikaments zu entscheiden.
Allerdings ist der Prozess recht aufwendig. Deshalb nur etwa 1 von 10 Medikamenten Nur wenige Medikamente, die in klinische Studien eintreten, erreichen jemals die Zulassung. Diese Zahl verdeutlicht die Herausforderungen dieses Weges und unterstreicht die bedeutende Rolle des Zulassungsantrags.
Eine Geheimhaltungsvereinbarung fungiert wie ein vollständiger “"Geschichte"” des Arzneimittels. Es enthält:
- Alle Ergebnisse klinischer Studien
- Vollständige Daten zu Sicherheit und Wirksamkeit
- Einzelheiten zur Herstellung des Medikaments
- Informationen zur Qualitätskontrolle und Stabilität
- Verpackung, Etikettierung und Gebrauchsanweisung
Die FDA prüft diese Informationen, um zu bestätigen, dass der Nutzen die Risiken überwiegt. Darüber hinaus beweist sie, dass der Herstellungsprozess ein sicheres und zuverlässiges Produkt gewährleistet.
Zweck eines NDA in der pharmazeutischen Industrie
Der Hauptzweck eines NDA besteht darin, nachzuweisen, dass ein neues Arzneimittel sicher, wirksam und in gleichbleibend hoher Qualität hergestellt wird.
Die FDA nutzt diesen Antrag, um zu bestätigen, dass jeder Teil des Produkts, von den klinischen Daten bis zum Herstellungsprozess, strengen Standards entspricht. Einer der Hauptgründe für diese Anforderung ist die frühzeitige Erkennung von Problemen.
Ein NDA (New Drug Application) dient auch dem Schutz von Patienten vor fehlerhaften oder unsicheren Produkten. Er verschafft der FDA (Food and Drug Administration) einen klaren Überblick über die Herstellung des Arzneimittels und die Einhaltung der Vorschriften. pharmazeutische Ausrüstung und die dahinterstehenden Prozesse können in jeder Charge die gleiche Qualität gewährleisten.
Wesentliche Bestandteile einer Geheimhaltungsvereinbarung
Ein Zulassungsantrag für Arzneimittel (NDA) umfasst mehrere Hauptabschnitte, die der FDA ein umfassendes Bild der Wirksamkeit eines Arzneimittels vermitteln. Im Folgenden finden Sie die wichtigsten Bestandteile, die Sie kennen sollten, um diese Anforderung zu erfüllen.
1. Klinisches Datenpaket
Das klinische Datenpaket umfasst alle Ergebnisse der Phase-1-, Phase-2- und Phase-3-Studien. Diese Studien zeigen, wie sich das Medikament im menschlichen Körper verhält, seine Wirksamkeit und mögliche Risiken, die während der Behandlung auftreten können.
Dieser Abschnitt enthält außerdem sämtliche Sicherheitsbewertungen und detaillierte Berichte über alle unerwünschten Ereignisse, die aufgrund der neuen Rezeptur aufgetreten sind.
2. CMC (Chemie, Fertigung und Kontrollen)
CMC ist einer der wichtigsten Bestandteile eines NDA, da er genau erklärt, wie das Arzneimittel hergestellt wird. Er umfasst Informationen über die Wirkstoff (API), einschließlich seiner Qualitätsstandards.
Darüber hinaus beschreibt diese Komponente den Herstellungsprozess vom Rohmaterial bis zum fertigen Produkt. Mithilfe dieser Informationen kann die FDA feststellen, ob das Medikament in großem Maßstab gleichbleibend und sicher hergestellt werden kann.
3. Etikettierung und Verpackung
Dieser Abschnitt beschreibt das vorgeschlagene Etikett des Arzneimittels und alle dem Produkt beiliegenden Anweisungen. Dazu gehören Dosierungsrichtlinien, Anwendungshinweise, Warnhinweise und Sicherheitsinformationen.
Klare Kennzeichnung und Serialisierung Sie sind unerlässlich für die Patientensicherheit und gewährleisten, dass die medizinischen Fachkräfte genau wissen, wie das Medikament zu verabreichen ist. Die FDA prüft diese Inhalte sorgfältig, um ihre Richtigkeit sicherzustellen.
4. Administrative und rechtliche Anforderungen
Der letzte Teil eines NDA-Antrags in der pharmazeutischen Industrie umfasst wichtige rechtliche und administrative Dokumente. In diesem Schritt müssen Sie Patientenbescheinigungen, Zahlungsnachweise für Nutzergebühren und weitere erforderliche behördliche Dokumente vorlegen.
Dazu gehören auch Konformitätsformulare, die bestätigen, dass das Unternehmen alle FDA- und gesetzlichen Verpflichtungen erfüllt.
Wie funktioniert der NDA-Einreichungsprozess?
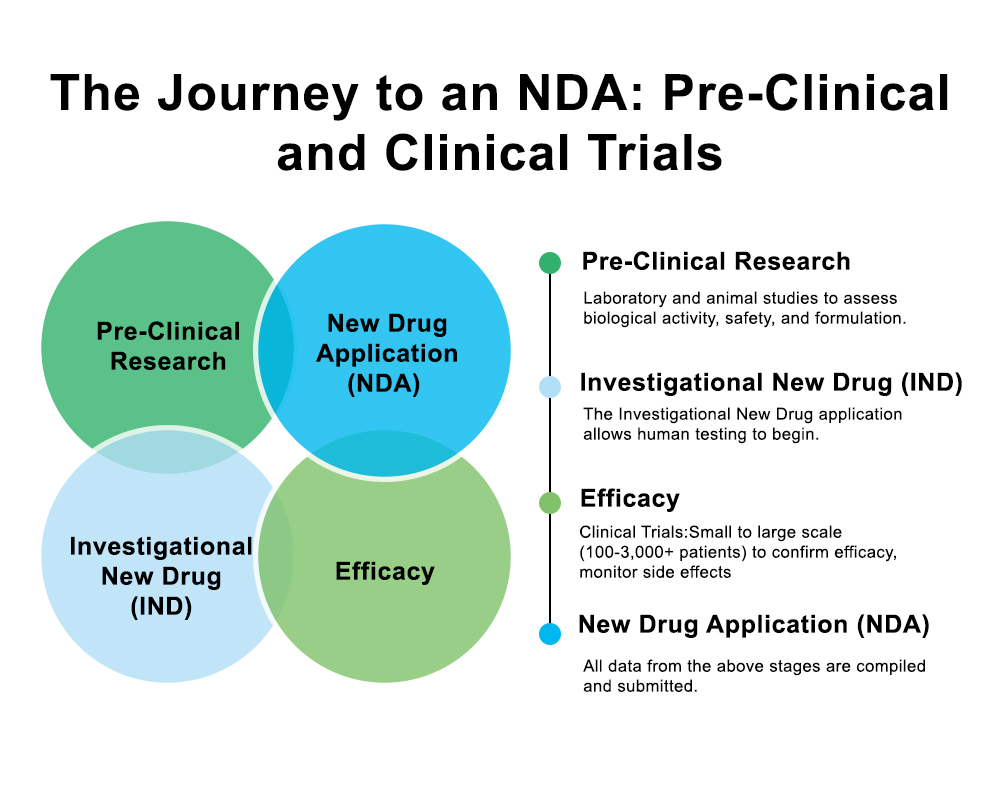
Die Einreichung eines NDA ist ein strukturierter Prozess, der dazu beiträgt, dass die FDA vollständige, genaue und qualitativ hochwertige Informationen erhält.
Folgende Schritte müssen Sie befolgen, um die Vorschriften einzuhalten:
1. Vorgespräch zur Einreichung des NDA-Antrags mit der FDA
Vor der Einreichung eines NDA-Antrags vereinbaren die meisten Unternehmen ein Vorgespräch. Dieses Gespräch gibt beiden Seiten die Möglichkeit, die finalen klinischen Ergebnisse, die Herstellungsdaten und etwaige verbleibende Bedenken zu besprechen.
Es trägt außerdem dazu bei, zu verdeutlichen, was die FDA von dem Antrag erwartet, was letztendlich das Risiko von Verzögerungen in der Zukunft verringert.
2. Erstellung des vollständigen Dokumentationspakets
Sobald der Plan abgestimmt ist, stellt das Unternehmen alle erforderlichen Daten zusammen, darunter klinische Ergebnisse, CMC-Informationen, Etikettenentwürfe und administrative Formulare.
Dieser Schritt erfordert die Abstimmung zwischen den Teams für Zulassung, Produktion, Qualitätssicherung und klinische Studien. Denken Sie immer daran: Ein gut organisiertes Paket erhöht die Wahrscheinlichkeit einer reibungslosen Prüfung.
3. Einreichung über das elektronische Portal der FDA
Der NDA-Antrag wird schließlich elektronisch über das Online-System der FDA eingereicht. ESG NextGen. Achten Sie darauf, dass die Datei den strengen Formatierungsregeln entspricht, damit die FDA sie bearbeiten und prüfen kann. Nach der Einreichung durchläuft der Antrag den internen Prüfprozess der FDA.
4. Ausführliche FDA-Überprüfung
Nach Einreichung des Prüfantrags beginnt die FDA mit einer detaillierten Auswertung aller Daten. Die Prüfer beurteilen die klinische Wirksamkeit, die Herstellungsqualität, die Sicherheitsinformationen und die Genauigkeit der Kennzeichnung.
In dieser Phase stellt die FDA häufig Fragen oder bittet um Klärung. Unternehmen müssen schnell und klar antworten, um Verzögerungen zu vermeiden.
5. Endgültige Entscheidung und Genehmigung
Wenn die Daten Sicherheit, Wirksamkeit und gleichbleibende Herstellung belegen, stellt die FDA eine Zulassungsbescheinigung aus. Stellt die FDA jedoch schwerwiegende Probleme fest, kann die Behörde eine Ablehnung aussprechen. Vollständiges Antwortschreiben mit einer Beschreibung dessen, was korrigiert werden muss.
Sobald Sie die Änderungen vorgenommen und die Genehmigung erhalten haben, bedeutet dies, dass Ihr Produkt nun in den USA vermarktet werden kann.
Wie lange dauert das NDA-Genehmigungsverfahren?
Die Dauer des Zulassungsverfahrens hängt von der Art der Prüfung ab, der das Arzneimittel unterzogen wird. Bei den meisten Produkten wendet die FDA ein Standardprüfungsverfahren an, das in der Regel etwa 10 bis 12 Monate ab Annahme des Antrags dauert.
Obwohl es Fälle gibt, in denen ein Medikament für eine beschleunigte Prüfung in Frage kommt, verkürzt sich die Zulassungszeit in diesem Fall fast um die Hälfte auf etwa sechs Monate. Eine beschleunigte Prüfung wird nur Medikamenten gewährt, die wesentliche Verbesserungen in der Behandlung versprechen oder schwerwiegende medizinische Bedürfnisse adressieren.
FAQs
1. Wie werden firmeneigene oder vertrauliche Informationen während der Prüfung der Geheimhaltungsvereinbarung geschützt?
Die FDA behandelt einen Großteil der klinischen und CMC-Daten in einem NDA als vertrauliche Geschäftsinformationen. Öffentliche Zusammenfassungen und die genehmigte Kennzeichnung werden zwar freigegeben, Rohdatensätze und firmeneigene Herstellungsdetails bleiben jedoch durch gesetzliche Vertraulichkeitsbestimmungen geschützt.
2. Was ist ein genehmigungsfähiges Schreiben bzw. ein vollständiges Antwortschreiben?
Kann die FDA die Zulassung in der eingereichten Form nicht erteilen, stellt sie ein vollständiges Antwortschreiben aus, in dem die Mängel und die erforderlichen Maßnahmen beschrieben werden. Ein Zulassungsschreiben legt die Bedingungen fest, die vor der Zulassung erfüllt sein müssen. Die Sponsoren reagieren darauf mit Änderungen, neuen Daten oder Vorschlägen zum Risikomanagement.
3. Wann spielen Patente und Exklusivität in einer Geheimhaltungsvereinbarung eine Rolle?
Sponsoren reichen Patentinformationen ein und können regulatorische Exklusivrechte anstreben, die den Markteintritt von Generika für einen bestimmten Zeitraum verzögern. Patentlisten im NDA unterstützen zudem die Patentzertifizierungsverfahren für spätere Generika-Antragsteller. Zu den Exklusivitätsarten gehören unter anderem die Exklusivität für neue chemische Substanzen, die Exklusivität für Kinderarzneimittel und die Exklusivität für Arzneimittel für seltene Erkrankungen.
Lassen Sie Ihre Geheimhaltungsvereinbarung nicht durch ungeeignete Maschinen verlieren
Die Einreichung eines Zulassungsantrags (NDA) in der pharmazeutischen Industrie ist ein wichtiger Meilenstein, doch die Zulassung hängt von mehr als nur klinischen Daten ab. Eines der wichtigsten Kriterien, die die FDA prüft, ist die Qualität der verwendeten Maschinen. Dies unterstreicht die Bedeutung zuverlässiger Lieferanten wie Finetech.
Unsere pharmazeutischen Maschinen sind darauf ausgelegt, eine zuverlässige und normkonforme Produktion in jedem Prozessschritt zu gewährleisten. Tatsächlich vertrauen Hersteller in über 60 Ländern und mehr als 500 Kunden auf Finetech, weil wir Anlagen liefern, die die für die FDA-Zulassung erforderliche technische Grundlage stärken.
Wenn Sie aufgrund von Maschinen festsitzen, Vereinbaren Sie ein kurzes Gespräch mit unseren Spezialisten. Heute!
Referenzen:
Alles über neue Medikamentenanträge (NDAs)
Vollständiger Leitfaden zu NDA vs. ANDA: Unterschiede, Verfahren und Anforderungen.
Was ist ein NDA (New Drug Application) und wie unterscheidet es sich von einer BLA?
Antrag auf Zulassung eines neuen Medikaments.
Copyright-Hinweis:
Es ist nicht gestattet, Inhalte dieser Website zu vervielfältigen, zu verändern, zu veröffentlichen, darzustellen, zu übermitteln oder in irgendeiner Weise zu verwerten oder solche Inhalte zum Aufbau von Datenbanken jeglicher Art zu verwenden, es sei denn, es liegt eine ausdrückliche schriftliche Genehmigung der Finetech Group vor. Für eine Genehmigung zur Nutzung des Inhalts wenden Sie sich bitte an: info@pharmamachinecn.com
Haftungsausschluss:
Die in diesem Artikel enthaltenen Informationen dienen lediglich der allgemeinen Information. Das Unternehmen übernimmt keine Garantie für die Richtigkeit, Relevanz, Aktualität oder Vollständigkeit der Informationen, und das Unternehmen übernimmt keine Verantwortung für Fehler oder Auslassungen im Inhalt dieses Artikels.