İçeriğe atla
İçeriğe atla Düzenleyici İşler (RA) bir ilaç şirketinin stratejik belkemiğidir. Bu disiplin, uluslararası kılavuzlara uyarak her ilacın kanıtlanmış, güvenilir ve emniyetli olmasını sağlar.
Özellikle sahte ve standart altı ürünler gibi sürekli tehditler göz önünde bulundurulduğunda, bu sürecin bütünlüğü kritik önem taşımaktadır. Yakın tarihli bir DSÖ raporuna göre US$30,5 milyar her yıl bu tehlikeli tıbbi ürünler için harcanmaktadır. Bu devasa rakam, küresel tedarik zincirlerindeki büyük bir güvenlik açığını vurgulamaktadır.
Düzenleyici İşler (RA), maksimum uyum ve koruma elde etmek için iki yönlü stratejik bir çaba olarak ortaya çıkmaktadır. Bilmek istiyorsanız bu makaleyi okumaya devam edin İlaçta ruhsatlandırma işlerinin ne olduğu, kimler tarafından yürütüldüğü ve işiniz için neden bu kadar kritik olduğu.
İlaç Sektöründe Düzenleyici İşler Nedir?
Ruhsatlandırma işleri (RA), her ilaç üretim organizasyonunda önemli bir departmandır. Şirket ile küresel ilaç düzenleme makamları arasında stratejik bir bağlantı görevi görür. Departmanın birincil rolü, her ilacın güvenlik, kalite ve terapötik performans için yasal ve bilimsel gereklilikleri karşılamasını sağlamaktır.
RA departmanı, güvenlik protokollerini denetlemenin ötesinde, laboratuvardan hastaya kadar olan tüm yolculuğu yönetir. Bu, klinik öncesi testlere ve klinik araştırma başvurularına rehberlik etmekten pazar izni için ruhsatlandırma dosyalarının hazırlanmasına kadar her şeye dahil oldukları anlamına gelir.
Düzenleyici İşler Uzmanları kimlerdir?
RA uzmanları, eczacılık veya tıp alanında geçmişleri olan ve genellikle özel Düzenleyici İşler (RA) sertifikalarına sahip uzmanlardır.
Bilimsel ürün verileri, düzenleyici yasalar ve uyumluluk yönergeleri hakkında güçlü bilgiye sahiptirler. Araştırma, üretim ve klinik ekipleriyle yakın işbirliği içinde çalışan RA uzmanları, uyumluluğun korunmasına ve pazar onaylarının güvence altına alınmasına yardımcı olur.
Temel hedefleri, farmasötik ürünlerin WHO ve ABD FDA gibi küresel düzenleyici otoriteler tarafından belirlenen standartları karşılamasını sağlamaktır.
Kilit Küresel İlaç Düzenleme Otoriteleri
İlaç düzenleme kurumları, bir ilacın kamu kullanımına uygunluğunu onaylayan ulusal ve uluslararası kuruluşlardır. İlaçlar da dahil olmak üzere her ilacın kamu kullanımına uygun olmasını sağlamak için küresel standartlar belirlerler. i̇laç üreti̇mi̇ süreci, kalite standartlarını karşılar ve güvenli ve etkilidir.
Düzenleyici İşlerin (RA) birincil rolü, şirket ile bu küresel ilaç düzenleme otoriteleri arasında temel irtibat ve tercüman olarak hareket etmektir.
Dünyanın en etkili ajanslarından bazılarına ve nasıl çalıştıklarına bir göz atalım.
1. Dünya Sağlık Örgütü (WHO)
The Dünya Sağlık Örgütü (DSÖ) farmasötikler için küresel normlar oluşturan ve teşvik eden uzmanlaşmış bir Birleşmiş Milletler kuruluşudur. Temel rolü, tüm ilaçların yüksek kaliteli, güvenli olmasını ve amaçlanan etkiyi göstermesini sağlamaktır. DSÖ aşağıdakiler gibi kritik standartlar belirler İyi Üretim Uygulamaları (GMP), Ulusal makamların dünya çapındaki ilaç üretim tesislerini denetlemek ve belgelendirmek için kullandıkları.
2. ABD Gıda ve İlaç İdaresi (FDA)
ABD. Gıda ve İlaç İdaresi (FDA) Amerika Birleşik Devletleri'nde halk sağlığını korumaktan sorumlu federal düzenleyici kurumdur. Yetki alanı beşeri ve veteriner ilaçları, aşılar, tıbbi cihazlar, gıda ve tütünü kapsamaktadır. FDA ürünleri düzenler, gerekli pazar onaylarını verir ve satılan tüm ürünlerin güvenli, güvenilir ve federal yasalara uygun olmasını sağlamak için harekete geçer.
3. Avrupa İlaç Ajansı (EMA)
The Avrupa İlaç Ajansı (EMA) beşeri ve veteriner ilaçlarının güvenliğinin izlenmesi ve denetlenmesinden sorumlu Avrupa Birliği kurumudur. Görevleri, endüstriye bilimsel tavsiyelerde bulunmak, yenilikçiliği teşvik etmek ve ürün yaşam döngüsü boyunca ilaç güvenliğini izlemektir.
4. Ulusal İlaç Düzenleme Kurumları
Ulusal ilaç düzenleme kurumları, kendi ülkelerinde satılan ilaçların ve ilgili ürünlerin kalitesini ve güvenliğini izleyen yerel devlet kurumlarıdır. Örnekler şunları içerir Sağlık Kanada, Japonya İlaç ve Tıbbi Cihaz Kurumu (PMDA), ve Birleşik Krallık İlaç ve Sağlık Ürünleri Düzenleme Kurumu (MHRA).
Düzenleyici İşleri Yönetmek: Sorumluluklar ve Süreçler
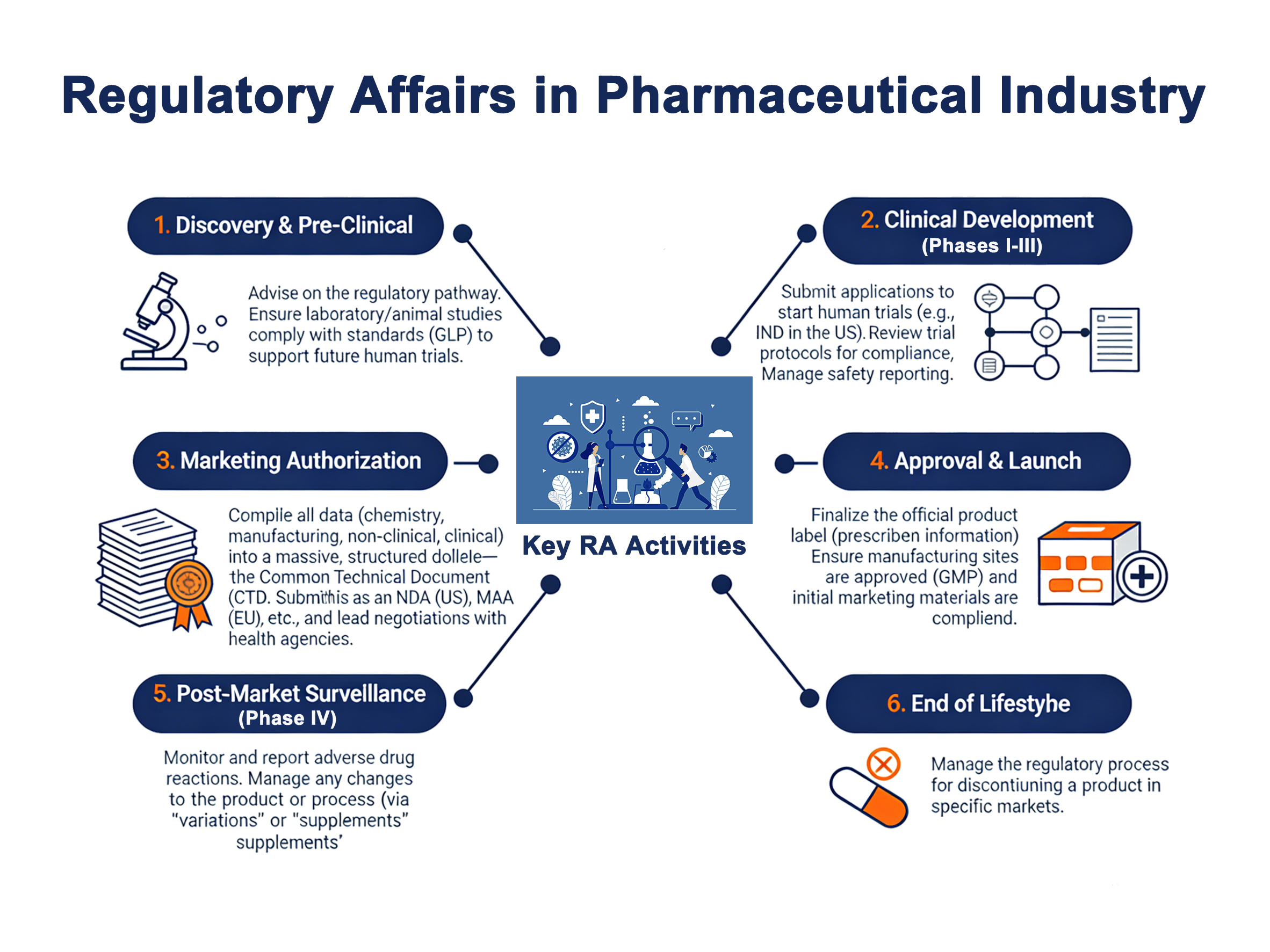
Düzenleyici işler uzmanları, bir ilaç kuruluşunun tüm yaşam döngüsü yönetiminin bel kemiğidir. API onayından ürünün nihai pazar iznine kadar, her profesyonel eylemin yasal olarak uyumlu olmasından sorumludurlar. İşte onların kapsamlı rollerini tanımlayan bazı temel sorumluluklar:
1. İlaç Etkin Maddesi Gönderimleri
İlaç üretimindeki ilk adım, API'ler için yasal uygunluğu sağlamaktır. Düzenleyici İşler uzmanları aşağıdakiler gibi temel belgeleri hazırlar ve sunar ilaç ana dosyaları (DMF'ler), boşluk analizi ve sürdürülebilirlik sertifikaları. Bu, kullandığınız API'lerin en yüksek kalitede olduğunu ve uluslararası standartlara uygun olarak üretildiğini kanıtlamak için gereklidir.
2. Uyum ve Kalite Güvencesi
İlaç üretiminde uyumluluk ve kalite güvencesi hasta güvenliği üzerinde doğrudan bir etkiye sahiptir. Düzenleyici işler uzmanları, tüm ürünlerin ve süreçlerin aşağıdakiler gibi katı uluslararası düzenlemelere uygun olduğunu doğrular İyi Üretim Uygulamaları (GMP) Ve İyi Klinik Uygulamaları (GCP).
Ayrıca, üretim sürecinin aşağıdakileri takip etmesini sağlarlar İlaç endüstrisindeki standart işletim prosedürleri (SOP'ler) ve makine ve tesislerin istenen kalite ve doğrulama standartlarını karşılaması.
3. Klinik Araştırmalar ve Gönderimler
RA uzmanları, kapsamlı düzenleyici stratejiler geliştirdikleri ve uyumlu klinik araştırma tasarımı konusunda tavsiyelerde bulundukları için klinik aşamanın ayrılmaz bir parçasıdır.
gibi belgelerin oluşturulmasından sorumludurlar. Araştırma Amaçlı Yeni İlaç (IND) başvurusu yaparak insan deneylerine başlar. Ekip ayrıca FDA gibi sağlık otoriteleri tarafından nihai ürün onayı için kapsamlı Yeni İlaç Başvurusunu (NDA) hazırlar.
4. Piyasa Onayı ve Yetkilendirme
Klinik araştırmalardan sonra, pazar iznini güvence altına almak da kritik bir görevdir. RA uzmanları, Ortak Teknik Belge (CTD), bilimsel araştırma, klinik dışı çalışma sonuçları ve kapsamlı klinik veriler dahil olmak üzere ilgili tüm bilgileri bir araya getirir.
Bu kapsamlı dosya, hem yerel hem de uluslararası makamların gereksinimlerini karşılamak ve şirketin kamu pazarına erişim durumunu oluşturmak için gereklidir.
5. Yetkili Makamlarla İrtibat ve İletişim
Mevzuat departmanı, şirketler ile Avrupa Komisyonu gibi kilit düzenleyici kurumlar arasındaki birincil temas noktasıdır. FDA ve EMA.
Bu profesyoneller basit yazışmalardan profesyonel müzakerelere kadar soruları yönetir, eksiklik mektuplarına yanıt verir ve ürünün gerekli unsurlarını tartışır.
6. Pazar Sonrası Yönetim ve Yaşam Döngüsü Uyumluluğu
İlaç onay aldıktan sonra, ruhsatlandırma ekipleri ürünün ömrünü pazar sonrası yönetim ve yaşam döngüsü uyumluluğu yoluyla yönetmek zorundadır. Önemli bir bileşen, hasta sağlığını korumak için sürekli güvenlik raporlaması ve advers olay verilerinin denetlenmesini içeren farmakovijilanstır.
Yaşam döngüsü uyumluluğu, onaylı üründe sonradan yapılan her değişiklik için karmaşık varyasyonların dosyalanmasını içerir. Bu gerekli değişiklikler, üretim yerinde bir değişiklik, revize edilmiş bir terapötik endikasyon veya nihai ürün etiketlemesinde bir güncelleme içerebilir.
Mevzuata Uyumsuzluk Riskleri
Farmasötik mevzuat uyumluluğunun tartışılmaz standartları vardır ve herhangi bir zayıf halka ciddi sonuçlara yol açabilir. İlaç yaşam döngüsü içinde gözetimdeki küçük bir ihmal bile ağır para cezalarına, ürün geri çağırmalarına ve cezalara yol açabilir. İşte böyle:
Risk No 1: Halk Sağlığı Etkisi
Halk, standart dışı veya sahte ilaçların ilk ve en savunmasız kurbanıdır. Sağlık Bakanlığı'na göre DSÖ, yaklaşık olarak her on ilaçtan biri düşük ve orta gelirli ülkelerde standartların altında veya tahrif edilmiş durumdadır. Bu tür ilaçlar, özellikle öksürük şurupları veya sıtma ilaçları gibi kritik ilaçlarda tedavi başarısızlığına, hastalığın ilerlemesine ve hatta ölüme yol açmaktadır.
Risk No 2: Geciken/İnkar Edilen Onaylar
Yeni bir ilacı piyasaya sürmek zaten on yıllık bir süreç. Genellikle 10 ila 15 yıl sürebilir İlk araştırmadan tüm klinik test aşamalarından geçtikten sonra nihai pazar mevcudiyetine kadar. Herhangi bir ciddi mevzuat ihlali veya dokümantasyon kusuru, pazar onayını daha da geciktirebilir veya tamamen reddedebilir.
Risk No 3: Para Cezaları ve Yasal İşlem
Sahte veya standartların altında ilaç üretmek ve satmak neredeyse tüm ülkelerde ağır bir suçtur. Örneğin, ABD'de sahte ilaç satmak ya da dağıtmak 10 yıla kadar hapis ve para cezasına yol açabilir. $250,000 bireyler için para cezası. Bu cezalar, hükümetlerin kamu sağlığını tehlikeye atan ihlalleri ele alma ciddiyetinin altını çizmektedir.
Ürününüzü Uyumluluk ve Yenilikle Güvence Altına Alın
İlaç sektöründe Düzenleyici İşlerin (RA) doğrudan sorumluluğunda olmasa da, üretim ekipmanlarının kalitesi uyumluluğun sürdürülmesinde çok önemli bir rol oynar.
Güvenilir, onaylanmış ve bakımı iyi yapılmış ekipman olmadan, en özenli uyum ve kalite çabaları bile başarısız olabilir. Bu nedenle, bir RA işe almanın yanı sıra, yüksek kaliteli, uyumlu üretim sistemlerine yatırım yapmak ve Finetech gibi güvenilir ekipman tedarikçileriyle ortaklık kurmak çok önemlidir.
Şu anda Finetech, gelişmiş teknik çözümler sunmanın ötesine geçiyoruz. Kapsamlı yerinde eğitimle ekibinizi güçlendiriyor ve çıktıyı en üst düzeye çıkarmanıza, uyumluluğu sürdürmenize ve uzun vadeli üretim mükemmelliğine ulaşmanıza yardımcı oluyoruz. Finetech ile bugün iletişime geçin ve uyumlu, yenilikçi üretimde geleceğinizi güvence altına alın.