Aller au contenu
Aller au contenu La phase de demande d'autorisation de mise sur le marché (AMM) est souvent celle où les équipes pharmaceutiques ressentent le plus de pression. C'est à ce moment que des années de développement, d'essais et d'investissement sont officiellement examinées par la FDA.
En termes simples, une demande d'autorisation de mise sur le marché (AMM) dans le secteur pharmaceutique est le document qu'une entreprise soumet à la FDA pour solliciter l'approbation d'un nouveau médicament. Il s'agit d'une étape cruciale dans tout processus de développement de médicament.
Ainsi, si vous développez une nouvelle formulation ou vous préparez à augmenter votre production, vous devez comprendre le processus de soumission d'une demande d'autorisation de mise sur le marché (AMM). C'est précisément ce sur quoi nous allons nous concentrer aujourd'hui. Entrons dans le vif du sujet.
Qu’est-ce qu’une NDA dans le secteur pharmaceutique ?

Un NDA est la demande officielle entreprise pharmaceutique Le demandeur soumet un dossier à la FDA pour obtenir l'autorisation de mise sur le marché d'un nouveau médicament. Il s'agit de l'étape finale du processus de développement d'un médicament et elle comprend tous les éléments nécessaires à la FDA pour déterminer si le médicament est sûr et efficace.
Cependant, le processus est assez exigeant. C'est pourquoi seulement environ 1 médicament sur 10 Parmi les médicaments entrant en phase d'essais cliniques, seuls quelques-uns obtiennent l'autorisation de mise sur le marché. Ce chiffre illustre les difficultés rencontrées et souligne le rôle crucial de la demande d'autorisation de mise sur le marché (AMM).
Un accord de non-divulgation (NDA) agit comme un accord complet “"histoire"” du médicament. Il contient :
- Tous les résultats des essais cliniques
- Données complètes sur la sécurité et l'efficacité
- Détails sur la fabrication du médicament
- Informations sur le contrôle de la qualité et la stabilité
- Emballage, étiquetage et instructions d'utilisation proposées
La FDA examine ces informations afin de confirmer que les avantages l'emportent sur les risques. De plus, cela prouve que le processus de fabrication permet de produire de manière constante un produit sûr et fiable.
Objectif d'une demande d'autorisation de mise sur le marché (AMM) dans le secteur pharmaceutique
L'objectif principal d'une demande d'autorisation de mise sur le marché (AMM) est de démontrer qu'un nouveau médicament est sûr, efficace et fabriqué de manière constante avec un haut niveau de qualité.
La FDA utilise cette application pour vérifier que chaque aspect du produit, des données cliniques à son processus de fabrication, respecte des normes strictes. L'une des principales raisons de cette exigence est de détecter les problèmes au plus tôt.
Une demande d'autorisation de mise sur le marché (AMM) vise également à protéger les patients contre les produits non conformes ou dangereux. Elle permet à la FDA d'avoir une vision claire du processus de fabrication du médicament et de sa conformité aux normes de sécurité. équipement pharmaceutique et les procédés sous-jacents permettent de garantir la même qualité pour chaque lot.
Éléments clés d'un accord de confidentialité
Une demande d'autorisation de mise sur le marché (AMM) pour un médicament comprend plusieurs sections importantes qui fournissent à la FDA une vision complète de l'efficacité du médicament. Voici les éléments à prendre en compte pour satisfaire à cette exigence.
1. Dossier de données cliniques
Le dossier de données cliniques comprend tous les résultats des essais de phases 1, 2 et 3. Ces études démontrent le comportement du médicament dans l'organisme, son efficacité et les risques potentiels pouvant survenir lors du traitement.
Cette section contient également toutes les évaluations de sécurité et les rapports détaillés de tous les événements indésirables survenus en raison de la nouvelle formulation.
2. CMC (Chimie, Fabrication et Contrôles)
La section CMC (Chimie, Fabrication et Contrôles) est l'une des parties les plus importantes d'une demande d'autorisation de mise sur le marché (AMM) car elle explique précisément comment le médicament est fabriqué. Elle couvre les informations relatives à la fabrication du médicament. principe actif pharmaceutique (API), y compris ses normes de qualité.
De plus, ce volet décrit le processus de fabrication, des matières premières au produit fini. Grâce à ces informations, la FDA peut déterminer si le médicament peut être produit de manière constante et sûre à grande échelle.
3. Étiquetage et emballage
Cette section décrit l'étiquetage proposé du médicament et toutes les instructions qui accompagneront le produit. Cela comprend les recommandations posologiques, le mode d'emploi, les mises en garde et les informations de sécurité.
Étiquetage clair et sérialisation Ces informations sont essentielles à la sécurité des patients et garantissent que les professionnels de santé savent précisément comment administrer le médicament. La FDA examine attentivement ce contenu afin d'en assurer l'exactitude.
4. Exigences administratives et légales
La dernière étape d'une demande d'autorisation de mise sur le marché (AMM) dans l'industrie pharmaceutique concerne les documents juridiques et administratifs essentiels. À ce stade, vous devez fournir les certificats des patients, les preuves de paiement des frais d'utilisation et autres documents réglementaires requis.
Il comprend également des formulaires de conformité confirmant que l'entreprise respecte toutes les obligations légales et celles de la FDA.
Comment fonctionne le processus de soumission d'un accord de non-divulgation (NDA) ?
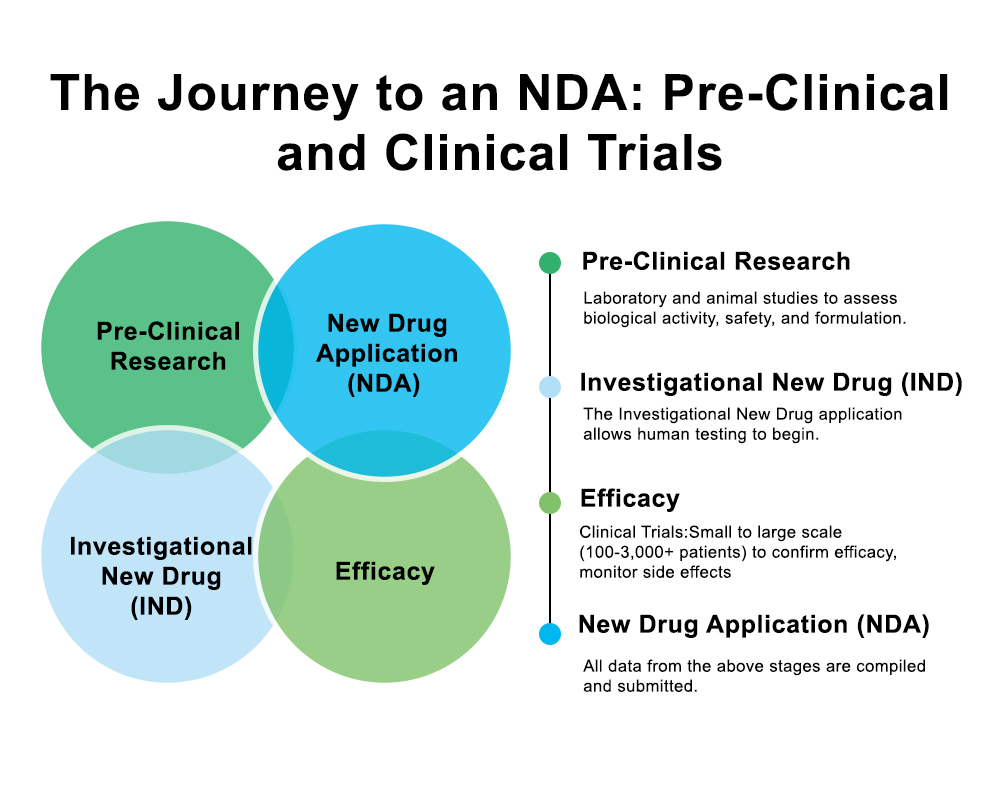
La soumission d'une demande d'autorisation de mise sur le marché (NDA) est un processus structuré qui contribue à garantir que la FDA reçoive des informations complètes, exactes et de haute qualité.
Voici les étapes à suivre pour être en conformité :
1. Réunion préalable à la demande d'autorisation de mise sur le marché (AMM) avec la FDA
Avant de soumettre une demande d'autorisation de mise sur le marché (AMM), la plupart des entreprises organisent une réunion préalable. Cette réunion permet aux deux parties d'examiner les résultats cliniques définitifs, les données de fabrication et les éventuelles questions en suspens.
Cela permet également de clarifier les attentes de la FDA concernant la demande, ce qui réduit à terme le risque de retards.
2. Préparation du dossier de documentation complet
Une fois le plan validé, l'entreprise compile toutes les données requises, y compris les résultats cliniques, les informations CMC, les ébauches d'étiquetage et les formulaires administratifs.
Cette étape exige une coordination entre les équipes réglementaires, de production, de qualité et cliniques. N'oubliez pas qu'un dossier bien organisé augmente les chances d'un examen sans encombre.
3. Soumission via le portail électronique de la FDA
La demande d'autorisation de mise sur le marché (NDA) est finalement soumise par voie électronique via le système en ligne de la FDA. ESG NextGen. Veillez à ce que le fichier respecte scrupuleusement les règles de formatage afin que la FDA puisse le traiter et l'examiner. Une fois soumise, la demande est intégrée au processus de sélection interne de la FDA.
4. Examen approfondi de la FDA
Une fois la demande d'examen déposée, la FDA procède à une évaluation détaillée de toutes les données. Les examinateurs évaluent les performances cliniques, la qualité de la fabrication, les informations relatives à la sécurité et l'exactitude de l'étiquetage.
Durant cette phase, la FDA envoie fréquemment des questions ou des demandes de précisions. Les entreprises doivent répondre rapidement et clairement afin d'éviter tout retard.
5. Décision finale et approbation
Si les données confirment l'innocuité, l'efficacité et la constance de la fabrication, la FDA délivre une lettre d'approbation. Toutefois, si la FDA identifie des problèmes majeurs, elle peut émettre un avis défavorable. Lettre de réponse complète décrire ce qui doit être corrigé.
Une fois les modifications apportées et l'approbation obtenue, votre produit pourra être commercialisé aux États-Unis.
Combien de temps dure le processus d'approbation d'une NDA ?
Le délai d'approbation d'une nouvelle demande d'autorisation de mise sur le marché (AMM) dépend du type d'examen auquel le médicament est soumis. Pour la plupart des produits, la FDA suit une procédure d'examen standard, qui dure généralement de 10 à 12 mois à compter de l'acceptation de la demande.
Bien que certains médicaments puissent bénéficier d'un examen prioritaire, le délai d'approbation est alors réduit de près de moitié à environ six mois. L'examen prioritaire est réservé aux médicaments qui apportent des améliorations majeures aux traitements existants ou qui répondent à des besoins médicaux importants.
FAQ
1. Comment les informations confidentielles ou exclusives sont-elles protégées lors de l'examen d'un accord de non-divulgation ?
La FDA considère une grande partie des données cliniques et CMC d'une demande d'autorisation de mise sur le marché (AMM) comme des informations commerciales confidentielles. Des résumés publics et l'étiquetage approuvé sont publiés, mais les données brutes et les détails de fabrication exclusifs restent protégés par les dispositions légales de confidentialité.
2. Qu’est-ce qu’une lettre approuvable ou une lettre de réponse complète ?
Si la FDA ne peut accorder l'approbation telle que soumise, elle émet une lettre de réponse complète décrivant les lacunes et les mesures requises. Une lettre d'approbation précise les conditions à remplir avant l'autorisation. Les promoteurs répondent en soumettant des amendements, de nouvelles données ou des propositions de gestion des risques.
3. Quand les brevets et l'exclusivité sont-ils importants dans un accord de non-divulgation ?
Les promoteurs soumettent des informations sur leurs brevets et peuvent solliciter des exclusivités réglementaires retardant l'arrivée des génériques sur le marché pendant des périodes définies. La liste des brevets figurant dans la demande d'autorisation de mise sur le marché (AMM) facilite également les procédures de certification des brevets pour les demandeurs de génériques ultérieurs. Parmi les types d'exclusivité, on peut citer l'exclusivité pour une nouvelle entité chimique, l'exclusivité pédiatrique et l'exclusivité pour un médicament orphelin.
Ne laissez pas votre accord de confidentialité vous échapper à cause de machines inadaptées.
Le dépôt d'une demande d'autorisation de mise sur le marché (AMM) dans l'industrie pharmaceutique est une étape majeure, mais son approbation dépend de bien plus que des seules données cliniques. L'un des critères les plus importants examinés par la FDA est la qualité des équipements utilisés. Cela souligne l'importance de s'appuyer sur des fournisseurs fiables comme Finetech.
Nos machines pharmaceutiques sont conçues pour garantir une production fiable et conforme aux normes à chaque étape du processus. De fait, des fabricants répartis dans plus de 60 pays et plus de 500 clients font confiance à Finetech car nous fournissons des équipements qui renforcent les bases techniques requises pour l'obtention de l'agrément de la FDA.
Si vous êtes bloqué à cause d'une machine, Prenez rendez-vous rapidement avec nos spécialistes. aujourd'hui!
Références :
Tout savoir sur les demandes de nouveaux médicaments (NDA)
Guide complet sur la NDA et l'ANDA : différences, processus et exigences.
Qu'est-ce qu'une NDA (New Drug Application) et en quoi diffère-t-elle d'une BLA ?
Demande d'autorisation de mise sur le marché d'un nouveau médicament.
Avis de droit d'auteur :
Vous ne pouvez pas reproduire, modifier, publier, afficher, transmettre ou exploiter de quelque manière que ce soit le contenu de ce site web, ni utiliser ce contenu pour constituer une base de données de quelque nature que ce soit, sans l'autorisation écrite expresse et préalable de Finetech Group. Pour obtenir l'autorisation d'utiliser le contenu, veuillez contacter : info@pharmamachinecn.com
Clause de non-responsabilité:
Les informations contenues dans cet article sont fournies à titre d'information générale uniquement. L'entreprise ne garantit pas l'exactitude, la pertinence, l'actualité ou l'exhaustivité des informations, et l'entreprise n'assume aucune responsabilité en cas d'erreur ou d'omission dans le contenu de cet article.